
Un año después de sorprender a los biólogos, AlphaFold ha cambiado la forma en la que los investigadores trabajan con las proteínas y ha puesto a DeepMind en un nuevo rumbo
En marzo de 2016, Demis Hassabis, CEO y cofundador de DeepMind, estaba en Seúl (Corea del Sur) viendo cómo la inteligencia artificial (IA) de su empresa hacía historia. AlphaGo, el programa informático entrenado para jugar al go, un juego de mesa tradicional, jugaba un partido de cinco juegos contra el destacado profesional coreano Lee Sedol, quien ostentaba entonces el segundo mayor número de victorias en campeonatos internacionales. Muchos consideran al go el juego de mesa más complejo del mundo. Se necesitan años para dominarlo.
Lee predijo que vencería a la IA de DeepMind con una victoria "aplastante", pero AlphaGo ganó 4-1, lo que dejó tan asombrados a los expertos en IA como a los de go y cambió la percepción de lo que la IA era capaz de hacer.
Pero mientras el equipo de DeepMind celebraba, Hassabis ya estaba pensando en un desafío aún mayor. Recuerda estar detrás del escenario con el entonces director del desarrollo de AlphaGo, David Silver, y decirle “ahora es el momento”.
Al ver cómo la IA de DeepMind jugaba a Go, Hassabis se dio cuenta de que la tecnología de su empresa estaba lista para enfrentarse a uno de los acertijos más importantes y complicados de la biología, para el cual los investigadores llevaban 50 años intentado dar una respuesta: predecir la estructura de las proteínas.
La estructura tridimensional de las proteínas determina cómo se comportan e interactúan en el cuerpo. Pero una gran cantidad de proteínas importantes tienen estructuras que los biólogos aún no conocen. Usar la IA para predecirlas con precisión ofrecería una herramienta valiosísima de cara a comprender las enfermedades, desde el cáncer hasta la COVID-19. Las proteínas son el objetivo principal para muchos medicamentos y un ingrediente clave en las nuevas terapias. Descubrir rápidamente sus estructuras aceleraría el desarrollo de nuevas terapias y vacunas.
En 2020, DeepMind, propiedad de Alphabet, presentó la IA AlphaFold2, que podía predecir la forma de las proteínas hasta el átomo más pequeño. "Es lo más complejo que hemos hecho nunca", asegura Hassabis.
El éxito de AlphaFold también es parte de una historia más grande e inicia un cambio de dirección para este laboratorio de IA. El foco de la empresa ha cambiado de los juegos a la ciencia, donde espera tener mayor impacto en el mundo real. Abordar problemas científicos es la culminación de lo que Hassabis se propuso lograr, y es por lo que querría ser reconocido. "Esta es la razón por la que creé DeepMind y, de hecho, por lo que trabajado durante toda mi carrera en la IA", confiesa.
Hassabis ha estado pensando en las proteínas de forma intermitente durante 25 años. Conoció ese problema cuando era estudiante en la Universidad de Cambridge (Reino Unido), en la década de 1990. "Un amigo mío estaba obsesionado con este problema", recuerda. "Lo mencionaba en cualquier ocasión, en el bar, jugando al billar, diciéndome que, si pudiéramos descifrar el plegamiento de proteínas, sería algo transformador para la biología. Su pasión siempre se quedó conmigo".
Este amigo es Tim Stevens, actualmente investigador de la Universidad de Cambridge especializado en las estructuras de las proteínas. "Las proteínas son las máquinas moleculares que hacen posible la vida en la Tierra", afirma Stevens.
Casi todo lo que hace nuestro cuerpo, lo hace con proteínas: digerir los alimentos, contraer los músculos, activar las neuronas, detectar la luz, potenciar las respuestas inmunitarias y mucho más. Por lo tanto, comprender el funcionamiento de las proteínas individuales es crucial para entender cómo funcionan los cuerpos, qué pasa cuando no responden y cómo repararlos.
Una proteína se compone de una cadena de aminoácidos que las fuerzas químicas repliegan con muchos giros, formando bucles complejos. Lo que hace una proteína está determinado por su forma tridimensional resultante. Por ejemplo, la hemoglobina (la proteína que transporta el oxígeno por todo el cuerpo y le da a la sangre su color rojo) tiene la forma de una pequeña bolsa que le permite recoger moléculas de oxígeno en los pulmones. La estructura de la proteína espiga del SARS-CoV-2 permite que el virus se adhiera a nuestras células.
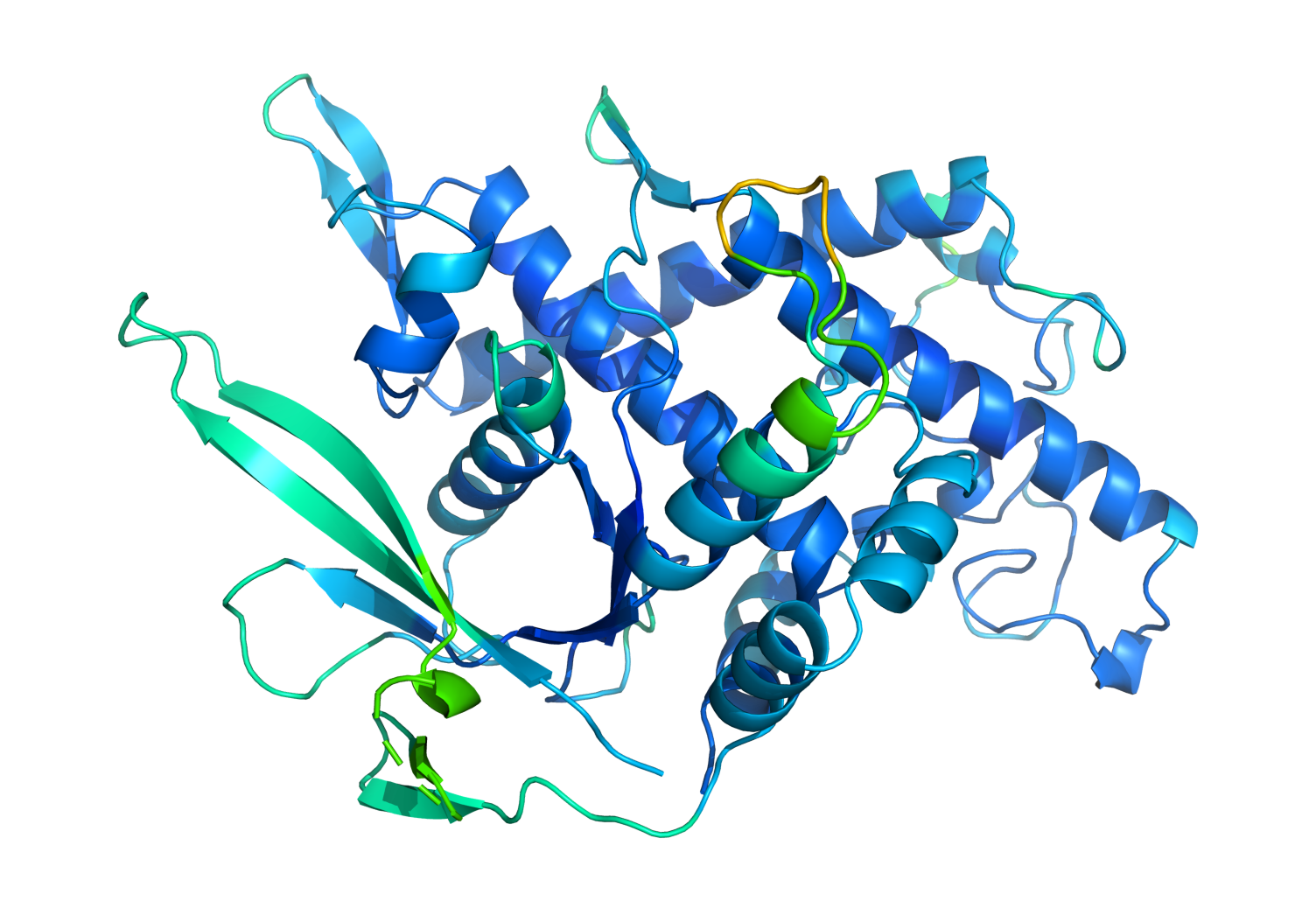
Foto: Un modelo generado por AlphaFold muestra cómo los aminoácidos se pliegan para formar una proteína. Créditos: Cortesía de Deepmind.
El problema consiste en que resulta difícil descifrar a partir de la cadena de aminoácidos la estructura de una proteína, y por lo tanto su función. Una cadena desplegada puede tomar 10 elevado a 300 formas posibles, un número del orden de todos los movimientos posibles en el go.
Predecir esta estructura en un laboratorio, utilizando técnicas como la cristalografía de rayos X, es un trabajo minucioso. Se han realizado doctorados completos dedicados a los plegamientos de una sola proteína. La competición CASP (Evaluación crítica de las técnicas para la predicción de la estructura de proteínas) se creó en 1994 para acelerar la investigación al enfrentar cada dos años los métodos de predicción computarizados entre sí. Pero ninguna técnica nunca estuvo tan cerca de igualar la precisión del trabajo de laboratorio. En 2016, el progreso llevaba una década estancado.
A los pocos meses del éxito de AlphaGo en 2016, DeepMind contrató a un puñado de biólogos y creó un pequeño equipo multidisciplinar para abordar el plegamiento de proteínas. El primer ejemplo de lo que hacían se mostró en 2018, cuando DeepMind ganó CASP 13, superando a otras técnicas por un gran margen. Pero más allá del mundo de la biología, pocos le prestaron atención.
Eso cambió cuando dos años después salió AlphaFold2. Ganó la competición CASP y fue la primera vez que una IA predijo la estructura de una proteína con una precisión que coincidía con la de los modelos generados en un laboratorio experimental, a menudo con márgenes de error del ancho de un átomo. Los biólogos quedaron atónitos por su buen rendimiento.
Al ver jugar a AlphaGo en Seúl, a Hassabis le recordó a un juego online llamado FoldIt, que había lanzado en 2008 un equipo dirigido por el conocido investigador de proteínas de la Universidad de Washington (EE. UU.) David Baker. FoldIt pedía a los jugadores que exploraran las estructuras de las proteínas, representadas como imágenes 3D en sus pantallas, plegándolas de diferentes formas. Los investigadores detrás del juego esperaban que, con mucha gente jugando, pudieran surgir algunos datos sobre las probables formas de ciertas proteínas. Aquello funcionó y los jugadores de FoldIt incluso contribuyeron a un puñado de nuevos descubrimientos.
"Si podemos imitar la máxima expresión de la intuición que es el 'go', ¿por qué no usarla para mapear las proteínas?"
Hassabis jugó ese juego cuando hacía su posdoctorado en el MIT a sus veintitantos años. Le llamó la atención la forma en la que la intuición humana básica podía conducir a avances reales, ya sea haciendo un movimiento en go o encontrando una nueva configuración en FoldIt.
"Me di cuenta de que lo que realmente habíamos conseguido con AlphaGo era imitar la intuición de los increíbles maestros de go", recuerda Hassabis. "Si podíamos imitar la máxima expresión de la intuición en el go, ¿por qué no podíamos usarla para mapear las proteínas?"
Los dos problemas no eran tan diferentes, en cierto modo. Al igual que go, el plegamiento de proteínas es un problema con una complejidad combinatoria tan grande que los métodos computacionales de fuerza bruta no lo pueden resolver. Otra cosa que go y el plegamiento de proteínas tienen en común es la disponibilidad de muchos datos sobre cómo se podría resolver el problema. AlphaGo usó el historial interminable de sus propios juegos anteriores; AlphaFold utilizó las existentes estructuras de proteínas de la base de datos internacional Protein Data Bank, de las estructuras resueltas que los biólogos han estado completando durante décadas.
AlphaFold2 utiliza las redes de atención, una técnica estándar de aprendizaje profundo que permite que la IA se centre en algunas partes específicas de sus datos de entrada. Esta tecnología respalda los modelos de lenguaje como GPT-3, donde dirige la red neuronal a las palabras relevantes en una frase. De manera similar, AlphaFold2 se dirige a los aminoácidos relevantes en una secuencia, como los pares que podrían estar juntos en una estructura plegada. " AlphaFold2 arrasó en la competición CASP gracias a que reunía cosas en las que los biólogos habían estado trabajando durante décadas y las dominaba mediante la IA", resalta Stevens.
Durante el año pasado, AlphaFold2 empezó a crear impacto. DeepMind ha publicado una descripción detallada de cómo funciona el sistema y ha hecho público el código fuente. También ha creado una base de datos pública con el Instituto Europeo de Bioinformática que se está llenando con las nuevas estructuras de proteínas tal y como las predice la IA. La base de datos actualmente tiene alrededor de 800.000 entradas, y DeepMind afirma que en el próximo año agregará más de 100 millones, casi todas las proteínas conocidas por la ciencia.
Muchos investigadores aún no comprenden completamente lo que ha hecho DeepMind, señala la directora científica de la empresa de descubrimiento de fármacos con IA de Reino Unido Exscientia y directora del laboratorio de informática de proteínas en la Universidad de Oxford (Reino Unido), Charlotte Deane, que también fue uno de los revisores del artículo que DeepMind publicó sobre AlphaFold en la revista científica Nature el año pasado. "Ha cambiado las preguntas que se pueden plantear", destaca Deane.
Varios equipos de todo el mundo han empezado a usar AlphaFold en sus trabajos sobre la resistencia a los antibióticos, el cáncer, la COVID-19, etc. El profesor y biólogo estructural del Fox Chase Cancer Center de Filadelfia (EE. UU.) Roland Dunbrack es uno de los primeros en adoptar esta tecnología. Dunbrack dirige un equipo que lleva años usando ordenadores para predecir las estructuras de proteínas. Luego, otros equipos en el laboratorio utilizan estas estructuras para sus experimentos.
AlphaFold ha introducido un nivel de precisión sin precedentes en el trabajo de Dunbrack. "Es lo suficientemente preciso para realizar evaluaciones biológicas e interpretar las mutaciones en un gen del cáncer", opina. "Ya intentábamos hacer eso con modelos generados por ordenador, pero a menudo nos equivocábamos".
Dunbrack ya puede tener más confianza en lo que les da a sus colegas. "Antes me ponía muy nervioso, me preocupaba que volvieran y me dijeran: desperdiciamos todo este dinero y tu modelo era terrible, no funcionó", bromea.
AlphaFold aún comete errores, pero cuando funciona bien, puede ser difícil diferenciar entre sus predicciones y una estructura generada en el laboratorio, resalta Dunbrack que realiza las predicciones de AlphaFold en la plataforma informática ColabFold, alojada en la Universidad de Harvard y que se ejecuta en las unidades de procesamiento gráfico de Google. "Todas las noches pongo una en marcha antes de irme a dormir, y tarda unas horas", explica Dunbrack.
"Es una herramienta muy útil que todos en mi laboratorio utilizan", afirma Kliment Verba, biólogo estructural de la Universidad de California en San Francisco. Verba se dedica principalmente al cáncer, pero en las primeras semanas de la pandemia de COVID-19, se unió a un consorcio de investigadores que estudiaban el virus SARS-CoV-2. Verba quería descubrir especialmente cómo sus proteínas secuestraban las proteínas del huésped.
Verba y sus colegas habían creado parte de la estructura de la proteína viral que les interesaba, pero les faltaba una pieza. Muchas proteínas tienen varios campos: secciones densamente plegadas, de cientos de aminoácidos de longitud, cada una de las cuales puede tener una función separada. Un campo podría unirse al ADN, otro a otra proteína, y así sucesivamente. "Son bestias de muchas cabezas", señala Dunbrack.
Estructuralmente, los campos son como los nudos de una cuerda, conectados por hebras sueltas que se enrollan y dan vueltas. El equipo de Verba había descubierto en la proteína que estaba estudiando la forma aproximada de la cuerda, pero no la estructura detallada de todos los nudos. Sin ese detalle, poco podían decir sobre su funcionamiento.
Sin embargo, se dieron cuenta de que esa proteína era una de las que DeepMind ya había pasado por AlphaFold y compartido online. La predicción de AlphaFold no fue perfecta; la curvatura de los giros no coincidía del todo. Pero tenía la forma de los cuatro campos de la proteína. Los investigadores tomaron las predicciones de AlphaFold para los campos y las compararon con la forma aproximada que ya tenían. Era sorprendentemente similar.
"Recuerdo ese momento de ver como encajaba todo", cuenta Verba. "Fue increíble. Éramos los únicos en el mundo con la estructura completa". Publicaron sus hallazgos poco después.
Verba cree que la fuerza de AlphaFold radica en encontrar las estructuras para las proteínas que aún no se han estudiado por completo: "Muchas de las proteínas que nos interesan se han analizado durante décadas. La gente se ha pasado la vida trabajando en ellas, por lo que tenemos una idea bastante clara de cómo son". Sin embargo, todavía queda mucho territorio desconocido.
Verba está interesado en las quinasas, por ejemplo. Las quinasas son enzimas que tienen un papel crucial en la regulación de la función normal de las células. Si dejan de funcionar correctamente, pueden causar cáncer. Solo alrededor de la mitad de las aproximadamente 500 quinasas del cuerpo humano se conocen bien; el resto se denomina el quinoma oscuro.
Los investigadores como Verba y Dunbrack tienen interés en desarrollar medicamentos contra el cáncer que se dirijan al quinoma. Pero es ahí donde se ven las limitaciones de AlphaFold.
Debido a que es costoso determinar la estructura de una proteína en el laboratorio, generalmente se lleva a cabo solo después de que la proteína haya sido elegida como candidata prometedora, lo que podría llevar meses en el proceso de descubrimiento de fármacos. Deane destaca que se espera que AlphaFold pueda revertir esa secuencia, volviendo más rápido el proceso. "Ahora puedo comenzar con la estructura: puedo identificar dónde tiene bolsillos en la superficie, lugares donde puedo unir las moléculas de los medicamentos", explica Deane.
"Muchas veces, estas pequeñas transformaciones son el punto decisivo de la función biológica"
No obstante, Deane reconoce que se necesita algo más que una estructura estática para comprender completamente cómo podrían interactuar un fármaco y una proteína. Las proteínas no se quedan quietas: sus estructuras pueden sufrir reconfiguraciones sutiles. "Muchas veces, estas pequeñas transformaciones son el punto decisivo de la función biológica", resalta Verba.
Es más, una proteína puede estar abierta a recibir un fármaco en un estado pero no en otros. Y a juzgar por lo que los investigadores están viendo hasta ahora, AlphaFold parece predecir el estado más común de estas estructuras, que puede no ser el estado importante para el desarrollo de fármacos.
Las proteínas también pueden cambiar de forma cuando los medicamentos se unen a ellas, lo que podría afectar el rendimiento del medicamento. En el peor de los casos, un fármaco que se une a una proteína puede tener efectos secundarios impredecibles en las proteínas contiguas, lo que podría incluso revertir la acción para la que fue diseñado el fármaco, por ejemplo, activar en vez de inhibir alguna función.
El jefe de IA molecular en ciencias de descubrimiento en AstraZeneca, Ola Engkvist, cree que las estructuras generadas por IA ayudarán a identificar los objetivos farmacológicos en un tiempo, pero todavía no. "Para ser transformacional, AlphaFold debe ir seguido de mejores métodos computacionales para comprender la dinámica de las proteínas y manejar complejos de proteínas más grandes", opina.
DeepMind planea abordar muchos de estos problemas en la próxima versión del programa. Una línea de trabajo es generar múltiples variaciones de la forma de una proteína para tratar de capturar su dinámica. La manera en la que se mueve una proteína se rige por una química y una física complejas, por lo que un modelo completo y en movimiento puede requerir alimentar AlphaFold con grandes cantidades de información adicional sobre este proceso. Un inconveniente de este enfoque podría ser que la información podría actuar como una restricción, degradando las capacidades predictivas de la herramienta.
El verano pasado, DeepMind lanzó AlphaFold Multimer, diseñado para predecir la estructura de los complejos de proteínas: las superestructuras hechas de múltiples proteínas agrupadas. Pero es mucho menos preciso que AlphaFold y propenso a errores más evidentes.
Los errores más tontos son una característica incluso de la mejor IA. AlphaGo cometió un error básico en el único juego que perdió ante Lee Sedol, señala Hassabis. "Se puede considerar un poco como un fallo de sistema", explica. "Pero el problema es que es un fallo en su conocimiento, no se puede corregir tan simplemente".
Eso se debe a que no se puede modificar muy fácilmente una red neuronal sin afectar fundamentalmente su funcionamiento. "Las correcciones codificadas dañan la capacidad de aprendizaje de la IA, ya que van en contra del propio aprendizaje".
En cambio, DeepMind está reuniendo los ejemplos de los errores más graves de AlphaFold y entrenándolo para manejarlos adecuadamente. Hassabis quiere que los investigadores entren en AlphaFold, para encontrar lo que no funciona, y compartir los resultados con su equipo para que puedan mejorar aún más el próximo AlphaFold.
Con AlphaFold, DeepMind está comenzando un nuevo capítulo. La empresa está invirtiendo en un equipo llamado AI for Science. Ha producido una avalancha de publicaciones en los últimos meses sobre distintos campos que van desde la predicción del tiempo hasta las matemáticas, la química cuántica y la fusión. Ninguna ha tenido el impacto de AlphaFold, pero la amplitud de la ambición es clara. "No tengo un librito de problemas que quiera abordar, pero tengo alguno en mente", lanza Hassabis.
AlphaFold también marca un nuevo capítulo para Hassabis. En noviembre, anunció que tenía un nuevo trabajo: compagina su liderazgo en DeepMind con el cargo de CEO en la start-up Isomorphic Labs, una nueva empresa de Alphabet que se centrará exclusivamente en llevar el poder de la IA a la biotecnología y a la medicina.
En esta etapa, Hassabis no quiere dar más detalles sobre lo que hará: “Acabamos de empezar, por lo que no hay mucho que decir. Básicamente, creo es posible hacer más cosas como AlphaFold, hay diferentes caminos hacia el descubrimiento de fármacos para los que la IA puede ser útil y no solo como una herramienta de análisis”.
En la entrada en la que anuncia en su blog el nacimiento de Isomorphic Labs, Hassabis escribe también que igual que las matemáticas resultaron ser el lenguaje adecuado de descripción para la física, la IA puede tener un papel similar para la biología.
Llevar a cabo este trabajo en su propia start-up facilita poder dedicarse al enfoque y a los recursos que necesita. "No tendría sentido contratar un montón de químicos en DeepMind", señala. Pero también es cierto que, mientras que DeepMind se ha limitado hasta ahora a la investigación pura (aparte de contribuir a los productos de Alphabet), la start-up buscará capitalizar lo que puede aportar a las grandes farmacéuticas.
"Puede ser similar a lo que hace DeepMind con Google", indica Hassabis. "Nuestra investigación se centra en cientos de productos de Google; casi todos los actuales productos de Google tienen algo de la tecnología de DeepMind. Isomorphic Labs podría ser algo como nuestra salida para el mundo real más allá de Google".
AlphaFold es para Hassabis un comienzo más que un punto final. "Vamos a ver un renacimiento en la ciencia, donde estas técnicas de IA se volverán cada vez más sofisticadas y se aplicarán a una amplia variedad de campos científicos", concluye. "A medida que crece la ola de la IA, hay más problemas que se vuelven abordables".
Inteligencia Artificial
La inteligencia artificial y los robots están transformando nuestra forma de trabajar y nuestro estilo de vida.
-
Al habla con la IA: el reto de lograr voces más diversas e inclusivas
La iniciativa Common Voice, de Mozilla, busca que las voces generadas por inteligencias artificiales sean más inclusivas y reflejen una mayor variedad de dialectos para asegurar que las particularidades de cada lugar y cultura se preserven frente al avance tecnológico

-
Estos robots aprendieron a superar obstáculos reales desde un entorno virtual gracias a la IA
Un nuevo sistema podría ayudar a entrenar robots usando exclusivamente mundos generados mediante inteligencia artificial

-
Por qué la IA podría comerle la tostada a la computación cuántica
Los rápidos avances en la aplicación de la inteligencia artificial a las simulaciones físicas y químicas hacen que algunos se pregunten si de verdad necesitamos ordenadores cuánticos
